
☰ Table of Contents
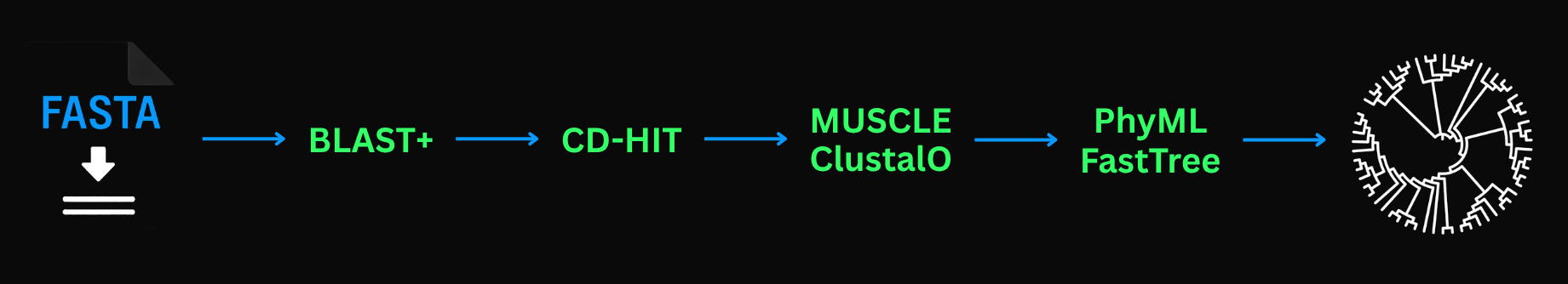
This is a course on the basics of working with protein sequences. The goal of this course is to teach you how to create phylogenetic trees of protein sequences to uncover their evolution and relationships in the command line from scratch, as well as how to use and understand all the tools needed on the way. It will cover the Linux command line, BLAST, sequence clustering, multiple sequence alignment and constructing and interpreting phylogenetic trees.
If you get stuck or have a question, let me know or ask ChatGPT / your LLM of choice. It's not perfect, but it's probably more accurate than me, as I have not yet fully escaped the title of a *Windows Wally. For legal reasons, this title is a joke.
💡 Pro bioinformatics tip: Protein alignments are prettier and also generally higher quality than DNA alignments, as the computer algorithm can compare and align 20 letters, rather than only 4.
1. Prerequisites
Windows on a laptop or PC and basic biology knowledge. Mac will work, setting up the CLI is probably easier. Even more so for Linux, but if you have Linux, why are you even here?
2. The Linux Command Line Interface
Most people use Windows or macOS. But heaps of computer people,
including and especially bioinformaticians, use Linux. Why? It is
open-source, efficient and flexible. So, getting your hands on the Linux
command line means getting quick access to an array of bioinformatics
tools. You might say, "But I can use BLAST on NCBI, why should I bother
using BLAST locally on my computer?". Because it is fast and flexible,
just like Linux. But more importantly, when your friends see you using
the
cd or ls commands and BLAST in the command
line, they might think you are a hacker or something. Oh, it also
unlocks a whole new level of power, letting you search custom databases,
tweak every step, and seamlessly integrate your analyses into complex
pipelines without any pesky server limitations.
Download Windows Subsystem for Linux (WSL):
https://learn.microsoft.com/en-us/windows/wsl/install
Once you have your command line running, we need to get to know it. Here are some ways to get a better grip on the commands.
Basic commands sheet:
Linux Commands Cheatsheet
YouTube tutorial (optional):
Linux Command Line Tutorial
You should be able to have the Linux command line open as well as regular programs in your Windows operating system, like Chrome.
⚠️ Wally Warning: Windows Wallies should be extra
careful now, as Windows is soft and forgiving. Linux is not. If you
remove a file with rm, it could be gone forever. If you
create a file that has the same name as an existing file, it just
vanishes. No questions asked.
Now that we can use the command line, let's download some bioinformatics software. Remember, usually if you ask ChatGPT how to download something in WSL, it will give you instructions or even the correct commands, as long as it is something simple. Or just google it. Probably safer.
To download all the programs, run these commands:
# BLAST
sudo apt install ncbi-blast+
# CD-HIT
sudo apt install cd-hit
# ClustalO
sudo apt install clustalo
# MUSCLE
sudo apt install muscle
# FastTree
sudo apt install fasttree
# PhyML
sudo apt install phymlWow, so quick! Make sure as many Windows Wallies see your screen while you are downloading, as they will now think you are 12.4% smarter than they previously did1. While we are at it, you may also want to download Jalview (the Windows version will do fine) and create an iTol account.
Jalview:
https://www.jalview.org/download/
iTol (Please take some time to admire my protein trees. This step is
critical.):
https://itol.embl.de/shared/maxstetina
Excellent. If you spent less than 1 minute admiring my trees, you have failed this course. For the successful admirers, let's move on to BLAST (it is such a basic bioinformatics tool, it's literally in the name, should be a piece of cake!).
3. BLAST (Basic Local Alignment Search Tool)
BLAST2 is an excellent tool to search for similar sequences to your sequence in a large database. It works with both nucleotide and protein sequences. This is how it works:
- Split the query sequence into "words"
- Generate similar "words" with similar AAs
- Search through the database with "words"
- Find identical or similar "words"
- Check for similarity in the vicinity "words"
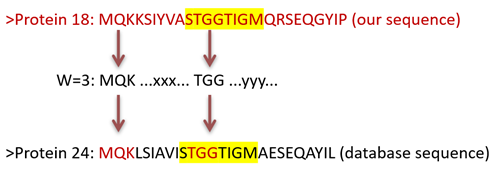
I'm surprised they didn't just call it Searching with "Words". Why does it work like this? Because it is fast! We can search through billions of sequences in hours or even minutes, which could take years with a bad algorithm.
So, we need two things. In our case, a protein sequence and a protein database. The sequence is easy, anything in a fasta format file. The second is a little harder, we need to download a protein database and prepare it for the BLAST search. Let's see how.
Let's download the manually annotated Swiss-Prot database from
UniProt:
https://www.uniprot.org/help/downloads
If we copy the link address for the Swiss-Prot fasta download in Chrome, we can then use the wget command to download directly into WSL.
wget {the copied link}
We then need to unzip it:
gunzip uniprot_sprot.fasta.gzWe should then have all the sequences in a file. Let's check the first couple of sequences:
head -n 300 uniprot_sprot.fasta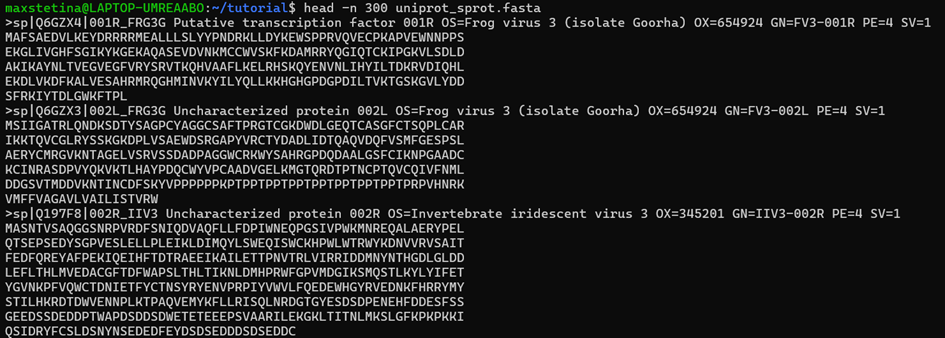
Congratulations, you have harnessed the power of biological data to your computer. Bioinformatics in action! Now, let's have a look at BLAST itself and prepare the database.
BLAST command options:
https://www.ncbi.nlm.nih.gov/books/NBK279684/
BLAST help in command line:
blastp -help
How to make a BLAST database out of any fasta sequence file:
https://www.ncbi.nlm.nih.gov/books/NBK569841/
Create the database:
makeblastdb -in uniprot_sprot.fasta -dbtype prot -out sprot -parse_seqids
💡 Pro bioinformatics tip: use
-parse_seqids so that later you can extract sequences
from the BLAST database using only sequence IDs. Thank me later.
Use this command to extract a sequence with an ID (optional):
blastdbcmd -db sprot -entry P00805Amazing, we have successfully built our first BLAST database. Remember, you can build a database like this from any fasta sequence file, including your special sequences. Now we can get down to BLASTing! This is what some BLAST outputs can look like in UniProt and the command line.
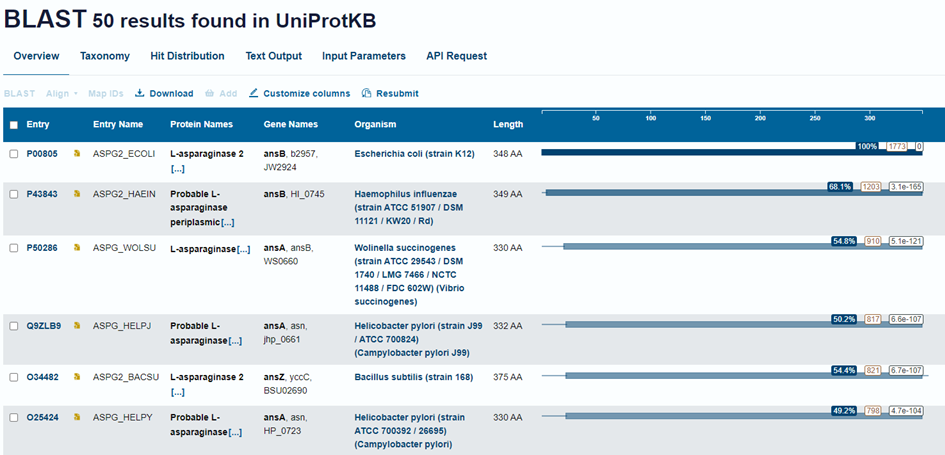
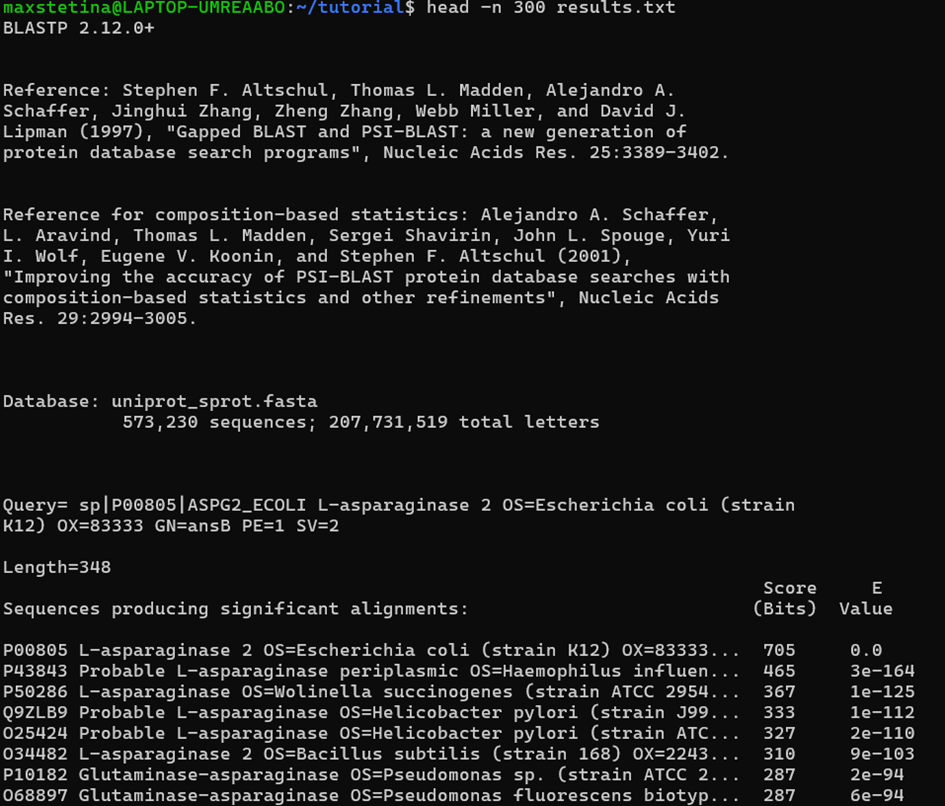
So, what do all these numbers mean?
-
Score (Bits): A measure of match quality; higher
means a better alignment and usually a more similar sequence.
- Calculated by BLAST using a substitution matrix, like BLOSUM62. Read about it here: https://en.wikipedia.org/wiki/BLOSUM.
🎉 Fun fact: After many years they found a mistake in how BLOSUM was calculated, so they fixed it. Naturally, the correct version worked worse than the one with the mistake3. Bioinformatics! Or just science in general!?
-
E-value (Expect or E-val): The number of hits
(sequences found) expected by chance; lower means more significant.
- If a hit has an E-val of 1, that sequence was probably found by chance and has no relevance to the query. An acceptable E-val could be considered to be 0.5, 0.1, 0.001 or even 0.00001 or lower, depending on the situation. It is the bioinformaticians job to "feel out" what a reasonable E-val is. That's you right now!
💡 Pro bioinformatics tip: Sometimes to get any results at all, you may need to use E-vals of ~0.5. But to have high confidence in your results, I would recommend E-vals on the lower end, like 0.00001. Keep in mind you cannot compare E-vals between different searches, as they are dependent on the database size, the length of the query and other factors.
- Identities: The exact matches between query and subject sequences.
- Positives: Matches that are similar in properties, not just identical.
- Gaps: Insertions or deletions introduced to optimize the alignment.
- +: Marks positions where amino acids are similar (Positives) but not identical.
We can use some of these numbers to define our search, they are great ways to cut-off what BLAST returns. So, let's start searching. Choose a protein sequence. I shall choose the Escherichia coli periplasmic asparaginase (P00805), an enzyme used in cancer therapy. We can copy the fasta sequence from UniProt (or for those actually paying attention, you just extracted it a few minutes ago) and save it in Linux with Nano.
Open the Nano editor while naming the new file and then copy and paste the sequence including the fasta header:
nano ASPG2_ECOLI.fastaKeyboard shortcuts to paste the sequence and close the editor:
right click
ctrl+x
y
enterYou should now have a file that looks like this.
Basic BLAST command:
blastp -query ASPG2_ECOLI.fasta -db sprot -out results.txtOur first BLAST search! The sequence found itself in the Swiss-Prot database, which is correct! It also naturally has the highest Score and lowest E-val. Good!
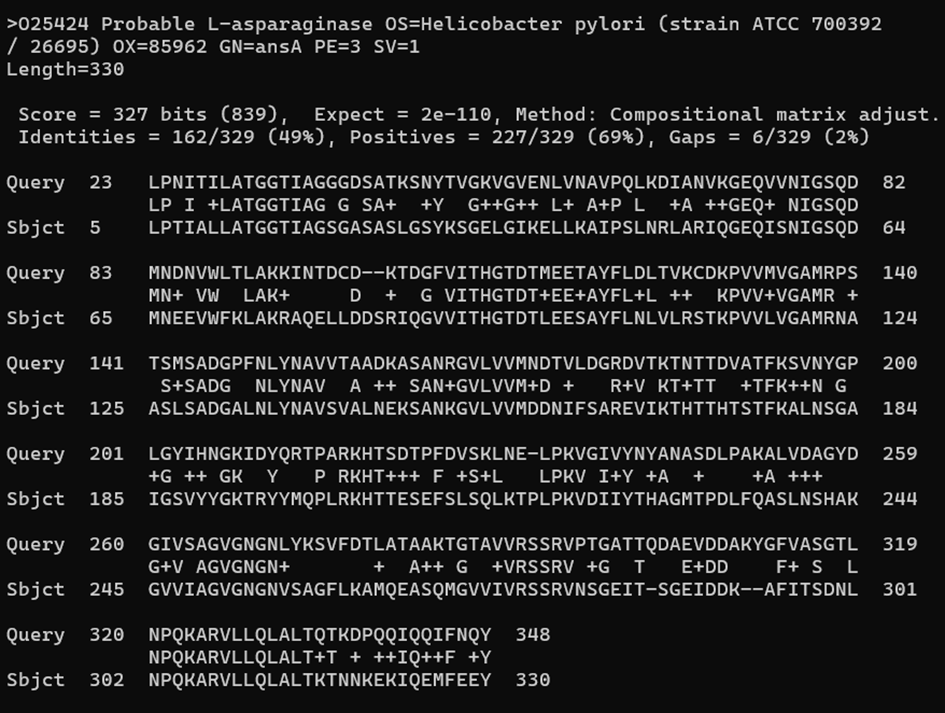
If we go lower, we find the pairwise alignments for each pair query-subject. Here is one from the middle and one from the bottom. The one in the middle looks great, but the one at the bottom has a very high E-val. It is also showing only a very short part of the subject sequence that is aligned. This similarity has been most likely found by chance!
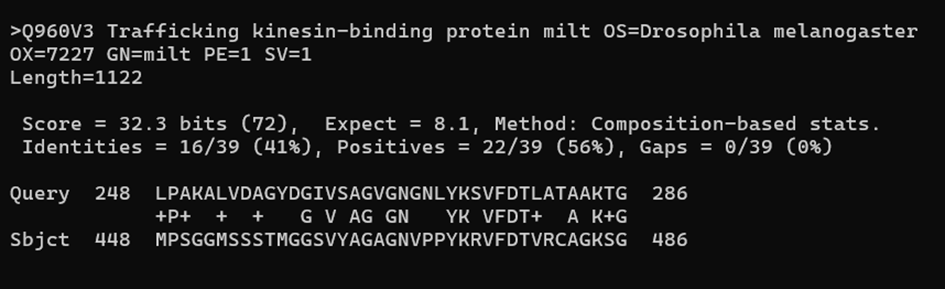
Having bad results mixed with our good results is not ideal. We can run the search again, but this time with a E-val cut-off of 0.001.
BLAST command with E-val cut-off:
blastp -query ASPG2_ECOLI.fasta -db sprot -out results.txt -evalue 0.001Now if I look at the results, towards the bottom we see a lot of Glutamyl-tRNA(Gln) amidotransferase subunit D proteins. What is this? Not an asparaginase? Was this found incorrectly or by chance? No! If we look at the alignments, the proteins are similar. If we go to UniProt and search for one of these sequences (Q9HJJ5) and go under Family & Domain, we will find this protein contains an Asparaginase/glutaminase domain, as well as some additional amino acids our E. coli asparaginase does not have. So, our BLAST search result is correct, but biology is complicated. This is something to always keep in mind when working with biological data.
💡 Pro bioinformatics tip: Use
-max_target_seqs {insert very big number} for larger
searches, as otherwise BLAST will limit the number of sequences found
and might not use your other cut-offs, like the E-val.
So now we have this default BLAST result file, which is great for understanding what is going on, but not so great for working with. If we check the BLAST manual, we can see that BLAST offers us several output formats, you can combine them to get BLAST to show you exactly what you want. In our case, we would like the sequence IDs, so we can then extract the sequences.
🪟 Wally bioinformatics tip: As far as I know, BLAST does not have an option to simply output the resulting sequences, which is a bit annoying. But what do I know?
BLAST command to output the sequence IDs:
blastp -query ASPG2_ECOLI.fasta -db sprot -out results.txt -evalue 0.001 -outfmt '6 sseqid'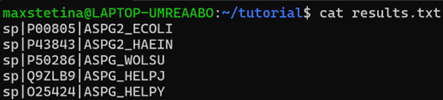
To get only the accession numbers, we can use a command like this:
cut -d'|' -f2 results.txt > accessions.txt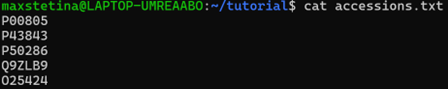
🪟 Wally bioinformatics tip: If you need a simple / not so simple command like this, or want to understand it, ChatGPT or your large language model of choice is happy to help.
If you use a different database, you may have to modify this formatting step or it might not be needed at all. Now we are ready to extract!
blastdbcmd -db sprot -entry_batch accessions.txt -out extracted_seqs.fasta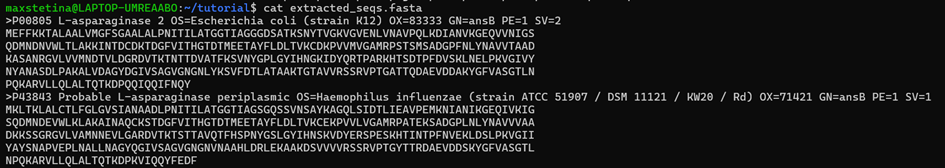
You have successfully employed BLAST, understood the results and prepared sequences in the fasta format for further analysis.
4. CD-HIT (Sequence Clustering)
"CD-HIT is very fast and can handle extremely large databases. CD-HIT helps to significantly reduce the computational and manual efforts in many sequence analysis tasks and aids in understanding the data structure and correct the bias within a dataset."4
In other words, you can take a large number of sequences and group them into clusters, depending on their similarity. You then work with a cluster representative, rather than all sequences that are very similar to each other. This is very useful when you have too many sequences, but in our case, we don't have that many sequences. We can use CD-HIT to remove extremely similar sequences, just so we lower redundancy in our future analysis.
💡 Pro bioinformatics tip: When working with huge amounts of sequences (hundreds of thousands or even more), we can use identity thresholds of ~60% or ~80% to significantly lower the number of sequences.
CD-HIT manual:
https://www.bioinformatics.org/cd-hit/cd-hit-user-guide
CD-HIT command line help:
cd-hit helpClustering sequences to an identity of 95%:
cd-hit -i extracted_seqs.fasta -o clustered_seqs.fasta -c 0.95 -n 5
We started with 74 sequences and finished with 58 clusters and 58
cluster representative sequences. This means that many sequences were
very similar to each other (within the clusters). We got two files, the
.clstr file, which shows us how the sequences were grouped.
The * signifies the representative sequences that we will be working
with further.
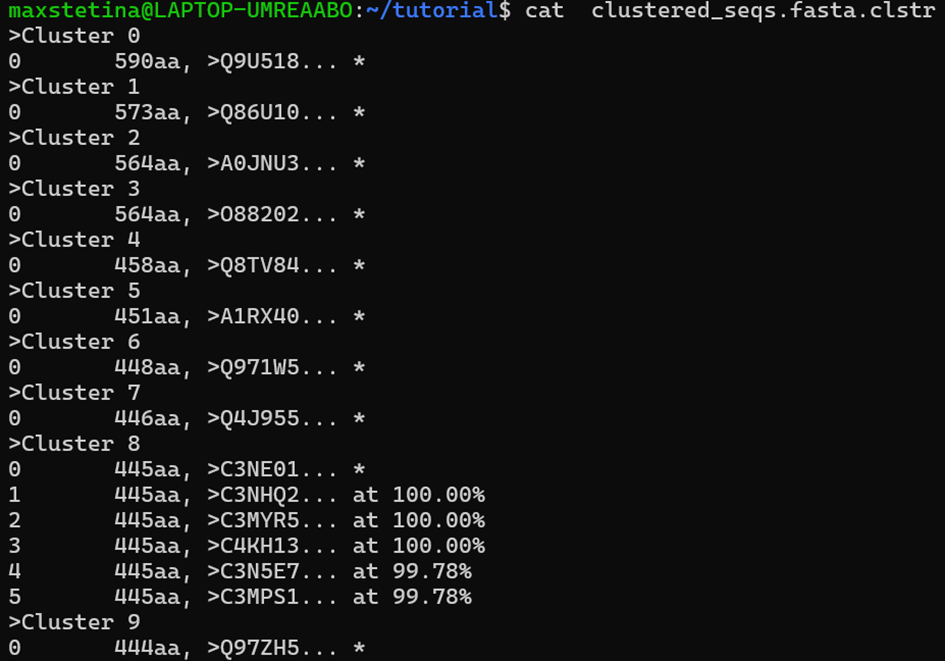
The second file are the fasta sequences of the representatives.
We can confirm that this file now only has 58 sequences, rather than the original 74, with the grep command:
grep -c '^>' clustered_seqs.fasta🪟 Wally bioinformatics tip: If you need a simple command like this, or want to understand it, ChatGPT or your large language model of choice is happy to help.
5. Multiple Sequence Alignment
"Multiple sequence alignment (MSA) is the process or the result of sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. These alignments are used to infer evolutionary relationships via phylogenetic analysis and can highlight homologous features between sequences."5
So, in contrast to BLAST we are comparing all the sequences to each other, rather than all the sequences (in a database) to a single query sequence.
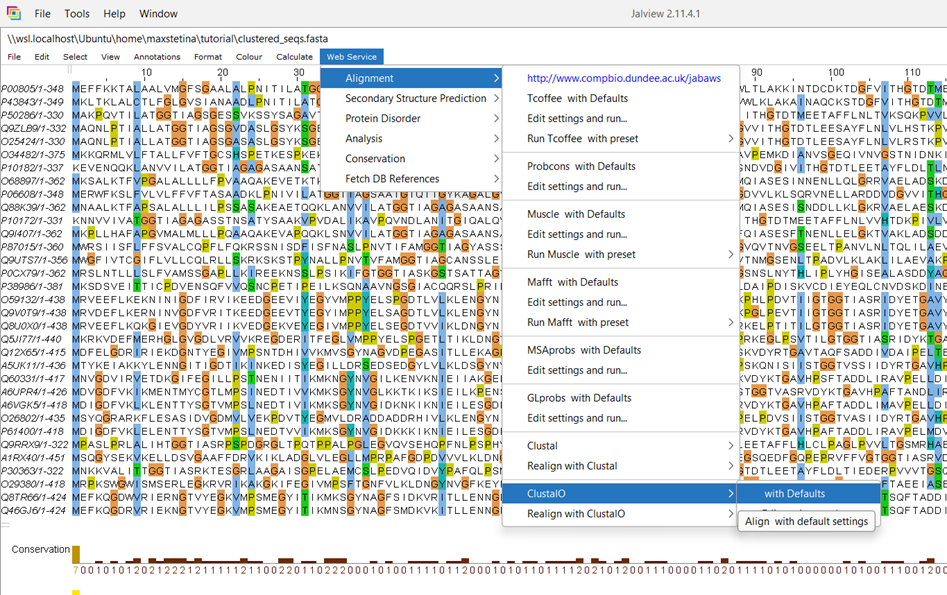
First, lets inspect and align the sequences in Jalview6.
You can either copy and paste them into a text box in Jalview or find where you are storing your sequences in the Windows file explorer.
\\wsl.localhost\Ubuntu\home\In Jalview, we have many algorithms we could use to align the sequences. ClustalO7 is a very commonly used one, as is MUSCLE8. Generally speaking, ClustalO is a bit faster, and MUSCLE is a bit more accurate. Try them both and see which alignment you prefer.
💡 Pro bioinformatics tip: As we are working with a relatively small number of sequences, these algorithms perform just fine. For working with large quantities, I like to use an algorithm called FAMSA, which is specialized at aligning huge protein families.
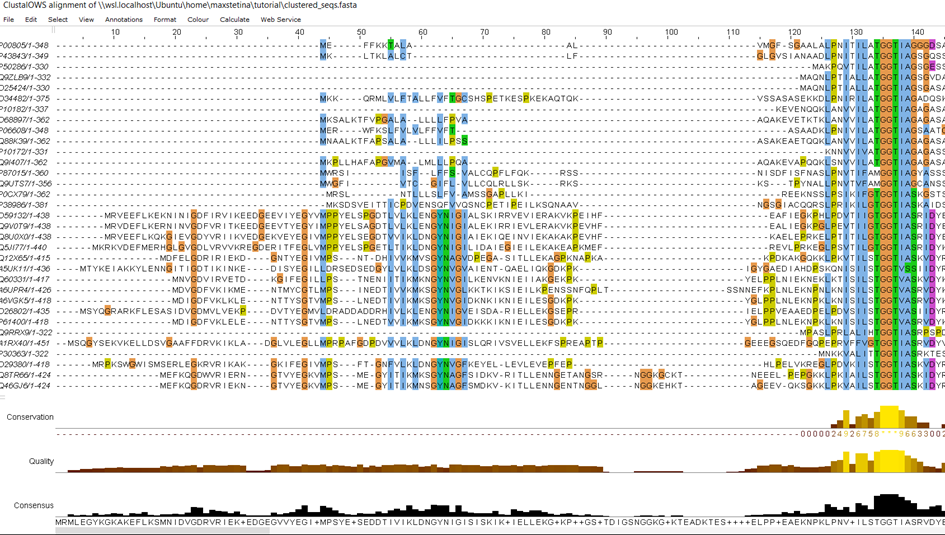
Because we are working with sequences from all the domains of life, the alignment has many gaps, but this is expected. You may have noticed the conserved motif of (T)GGT, which all of the sequences share. This is because this motif is in the active site of the enzyme and is needed for its activity, therefore, all the sequences have it! This is an indication our alignment is correct. If you feel you can do better than the algorithm, you can edit it in Jalview, which is sometimes necessary to get quality alignments.
💡 Pro bioinformatics tip: MSA algorithms work better when you give them sequences that are actually similar. We don't have to worry about this, as we got the sequences from a similarity search (BLAST) with a solid E-val. It's preferable you generally don't feed the MSA algorithms completely dissimilar sequences, as this will likely give you bad results.
But this is how a Windows Wally would align sequences. We can do it with cool commands, instead.
Instructions (Clustalo and MUSCLE):
https://open.bioqueue.org/home/knowledge/showKnowledge/sig/clustalo
https://www.drive5.com/muscle/muscle.html
Command line help:
clustalo --help
muscleExecute MSA:
clustalo -i clustered_seqs.fasta -o aligned_clustalo.fasta
muscle -in clustered_seqs.fasta -out aligned_muscle.fasta⁉️ Windows Wally rant: Yes, bioinformaticians have a hard time agreeing on how to format things. They tend to make programs with very slightly different input formats. Annoying.
As you may have noticed, aligned sequences are nothing more than sequences with gaps.
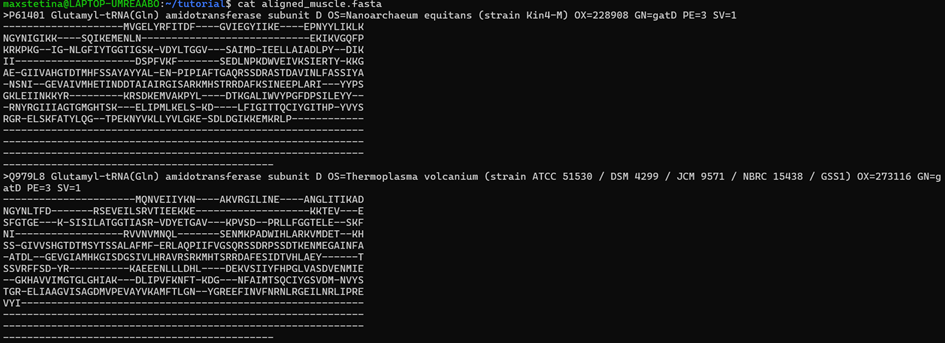
6. Constructing Phylogenetic Trees
"A phylogenetic tree, phylogeny or evolutionary tree is a graphical representation which shows the evolutionary history between a set of species or taxa during a specific time."9
Although phylogenetic trees are usually used to show the relationships between species, we can do the same for genes (or proteins). After all, phylogenetic trees of species are often constructed based on the combined analysis of several selected genes. So why not do the same for a single protein? Although we may not be strictly describing how the protein linearly "evolved", as proteins undergo convergent evolution and their genes horizontal transfer events. Nonetheless, we can use these trees to understand the similarities between individual protein sequences or groups of sequences and sometimes infer something about their evolution. This can help us better understand which proteins are more likely to have certain properties, as we shall see next.
As with MSA algorithms, we can choose from an array of phylogenetic tree constructing algorithms. For larger numbers of sequences, FastTree10 is a solid choice. For higher quality trees, PhyML11 is a solid option, but will take a lot longer even for our 58 sequences. You can combine the MSA and the tree algorithm as you wish. Choose the one you like the best.
FastTree and PhyML instructions
http://www.microbesonline.org/fasttree/
https://manpages.ubuntu.com/manpages/bionic/man1/phyml.1.html
Command line help:
fasttree
phyml --helpBuild a tree with FastTree and the MUSCLE alignment:
fasttree aligned_muscle.fasta > asparaginase_muscle_fasttreeRun ClustalO again to get an output format suitable for PhyML, --force will overwrite your previous alignment with the same name:
clustalo -i clustered_seqs.fasta -o aligned_clustalo.phy --outfmt=phylip --forceBuild a tree with PhyML:
phyml -i aligned_clustalo.phy -d aaThe result will be:
aligned_clustalo.phy_phyml_tree.txtWhile PhyML is running (may take several minutes), we can check out our first tree in iTol12.

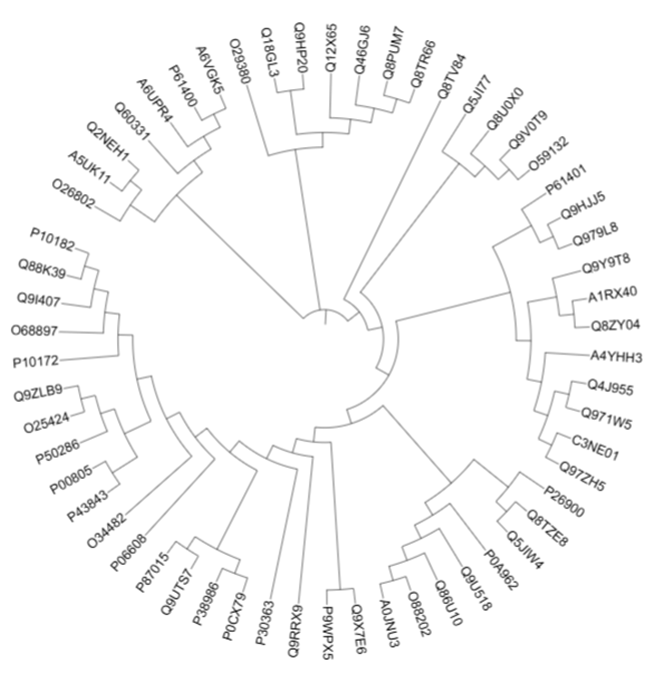
Interpreting a tree by ChatGPT:
1. Branches (Edges)
- Represent evolutionary relationships.
- The length often reflects the amount of change (e.g., number of substitutions).
2. Nodes
- Internal nodes represent common ancestors.
- Tips (leaves) are the protein sequences you provided.
3. Clades
- A group of proteins that share a common ancestor.
- Proteins in the same clade are typically more closely related in structure/function.
4. Branch Support Values
- Numbers on branches (from bootstrap or aLRT) indicate confidence in the grouping.
- >70% bootstrap or >0.9 aLRT is generally considered strong support.
The main point is, if the sequences are grouped together, they are more similar. If groups of sequences are further apart, they are less similar. We can also tell which sequence came first in evolution and which ones have recent or less recent common ancestors (if we are lucky).

I like the unrooted trees (see the control panel) and usually I like to root them at the midpoint. Yes, this sounds odd, but this allows us to copy sequence IDs from specific regions easier.
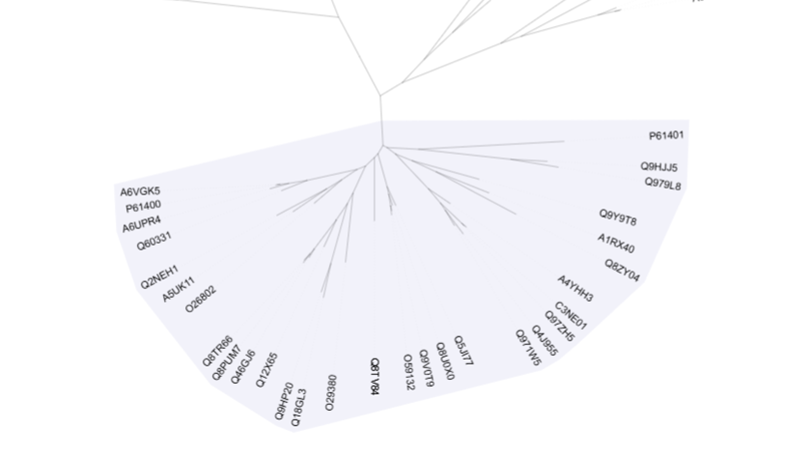
Here I have highlighted one of the regions that looks like a bundled group of sequences, further from the others. Perhaps it is a phylogenetic family and maybe the proteins have different properties to some of the other families. To check, we can left click on the branch leading to this area and copy the leaf labels.
To see more details, we can map the sequences with UniProt ID Mapping
by pasting them here:
https://www.uniprot.org/id-mapping
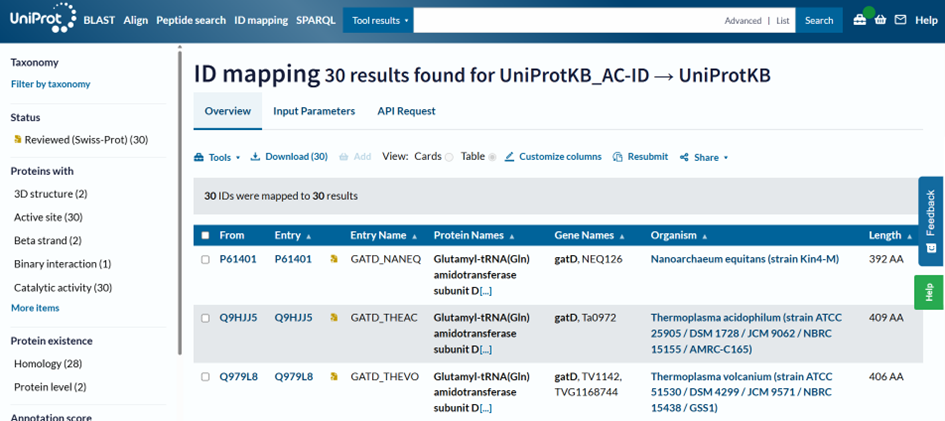
All the sequences in this area are Glutamyl-tRNA(Gln) amidotransferase subunit D proteins we talked about earlier! We have successfully grouped them together and understood that this branch of sequences is somehow different to the rest, and in this case, they also have a completely different activity, although their sequences are very similar. If we go back to Jalview, we may see that not only the asparaginase domain has some specificities, but they also have an additional domain at the N-terminus.
Let's check out the tree made by ClustalO and PhyML.

The results are very similar, but we can see in some cases FastTree tended to put sequences on individual branches, rather than group them on to shared ones. You can decide which one you like more. You can also inspect more branches to see what's going on and map more sequences to UniProt to better understand and interpret your results. iTol allows paying users to further annotate and colour the trees, but most of this can be done in your favourite graphic design tool, Microsoft Paint. Or any other.
Congratulations on building your first phylogenetic trees in the command line. Although what we did could have been achieved on the UniProt website, we looked cooler doing it. Also, now we can do it with greater control, automation, reproducibility, and the ability to handle large datasets without file size limits or timeouts—plus, it's faster, scriptable, works offline, and integrates seamlessly into larger bioinformatics pipelines.
If you found this tutorial useful or have a question feel free to let me know and please consider sharing it and/or citing:
Bioinformatics for Windows Wallies by Max Stetina (2025), licensed under CC BY-NC-SA 4.0,
available at: https://bioinformaticsforww.dyn.cloud.e-infra.cz/tutorial-1.htmlReferences
1 The Book of Undisputable Bioinformatics Facts by Max Stetina (not available for the public)
2 Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402.
3 Styczynski, M., Jensen, K., Rigoutsos, I. et al. (2008), "BLOSUM62 miscalculations improve search performance", Nat Biotechnol 26, 274–275. https://doi.org/10.1038/nbt0308-274
4 Weizhong Li's Group. (2022). CD-HIT. [Online]. Available:
https://sites.google.com/view/cd-hit/home
[Accessed: May 25, 2025]
Weizhong Li, Adam Godzik (2006), "Cd-hit: a fast program for
clustering and comparing large sets of protein or nucleotide
sequences", Bioinformatics, Volume 22, Issue 13, Pages 1658–1659.
https://doi.org/10.1093/bioinformatics/btl158
5 Wikipedia. (2024). Multiple sequence alignment. [Online]. Available: https://en.wikipedia.org/wiki/Multiple_sequence_alignment [Accessed: May 25, 2025]
6 Waterhouse, A.M., Procter, J.B., Martin, D.M.A, Clamp, M., Barton, G.J (2009), "Jalview version 2: A Multiple Sequence Alignment and Analysis Workbench", Bioinformatics 25 (9) 1189-1191. https://doi.org/10.1093/bioinformatics/btp033
7 Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011), "Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega", Mol Syst Biol. 7:539. https://doi.org/10.1038/msb.2011.75
8 Robert C. Edgar (2004), "MUSCLE: multiple sequence alignment with high accuracy and high throughput", Nucleic Acids Research, Volume 32, Issue 5, Pages 1792–1797. https://doi.org/10.1093/nar/gkh340
9 Wikipedia. (2025). Phylogenetic tree. [Online]. Available: https://en.wikipedia.org/wiki/Phylogenetic_tree [Accessed: May 25, 2025]
10 Price MN, Dehal PS, Arkin AP (2010), "FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments", PLOS ONE 5(3): e9490. https://doi.org/10.1371/journal.pone.0009490
11 Stéphane Guindon, Olivier Gascuel (2003), "A Simple,
Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum
Likelihood", Systematic Biology, Volume 52, Issue 5, Pages 696–704.
https://doi.org/10.1080/10635150390235520
12 Ivica Letunic, Peer Bork (2024), "Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool", Nucleic Acids Research, Volume 52, Issue W1, Pages W78–W82. https://doi.org/10.1093/nar/gkae268